EDEMA AGUDO DOS PULMÕESUnidade de Terapia
Intensiva — Disciplina de Emergências Clínicas — HC-FMUSP Unidade
Clínica de Emergência — Instituto do Coração (InCor) — HC-FMUSP
Endereço para correspondência:
Unidade Clínica de Emergência — InCor — Av. Dr. Enéas Carvalho de
Aguiar, 44 — CEP 05403-000 — São Paulo — SP
INTRODUÇÃO
O edema agudo dos pulmões representa uma das principais causas de
insuficiência e/ou desconforto respiratório que motivam a procura de
unidades de emergência ou terapia intensiva. Nos Estados Unidos, é a
causa mais freqüente, seguida das doenças brônquicas primárias e das
encefalopatias agudas decorrentes de acidentes vasculares cerebrais e
traumatismos cranioencefálicos( 1).
O edema pulmonar ocorre pelo desequilíbrio das forças de Starling,
podendo ocorrer aumento da pressão hidrostática capilar e/ou aumento
da permeabilidade dos capilares pulmonares(2, 3). Neste texto, daremos
destaque às condições que cursam com aumento da pressão hidrostática
pulmonar de origem cardiogênica.
FISIOPATOLOGIA
A disfunção cardíaca causa elevações na pressão venosa pulmonar, com
conseqüente aumento da pressão hidrostática nos capilares pulmonares e
da ultrafiltração do intravascular para o interstício pulmonar. O
interstício dos septos interalveolares tem pressão hidrostática
negativa, porém menos negativa que os espaços peribrônquicos,
decorrente da drenagem linfática ativa destes últimos e pela
ultra-estrutura do esqueleto pulmonar. As forças de tração que
resultam da expansão pulmonar e da ventilação são aplicadas
diretamente nos espaços peribrônquicos, e, a partir destes,
distribuídas para os outros componentes da estrutura pulmonar. A
resultante dessas forças leva ao fluxo unidirecional dos fluidos no
interstício pulmonar dos septos interalveolares, onde é coletado o
líquido ultrafiltrado dos capilares para os espaços peribrônquicos. É
nos espaços peribrônqicos que esse líquido é captado pelo sistema
linfático e devolvido para a circulação venosa sistêmica. O sistema
linfático tem propriedades como o sistema valvar, que permite apenas o
fluxo unidirecional. As paredes dos capilares são fixadas por meio de
fibras colágenas nos septos interalveolares; assim, durante a
inspiração, as paredes capilares são tracionadas em direções
centrífugas, originando pressão negativa em seu interior. Por outro
lado, durante a expiração, com o relaxamento das paredes capilares, a
pressão em seu interior torna-se positiva por causa da retração
elástica das paredes capilares e da contração dos periscitos
capilares. Esse jogo de variação de pressões durante o ciclo
respiratório aplicado a um sistema canalicular linfático valvado gera
um fluxo em direção ao sistema venoso(2, 3). Com esse mecanismo básico
de drenagem de líquidos do terceiro espaço pulmonar, fica lógico que,
quanto maior a freqüência respiratória e/ou a amplitude das
inspirações, maior será a drenagem linfática. Além disso, no
interstício pulmonar existem terminações nervosas com sensibilidade
química (quimiorreceptores) e mecânica (mecanorreceptores e
proprioceptores). Os mecanorreceptores (entre eles os receptores J ou
justa-alveolares) são capazes de perceber o aumento da pressão
hidrostática e/ou aumento do fluxo de líquidos. Esse estímulo é
conduzido pelas suas eferências, que provocam aumento da descarga
periódica do centro respiratório; isso resulta no aumento da
freqüência respiratória, provocando drenagem linfática maior pelo
mecanismo já descrito. Dessa maneira, forma-se o principal mecanismo
de defesa pulmonar contra o aumento de ultrafiltração capilar
pulmonar. Esse mecanismo permite o aumento do ultrafiltrado de 20 ml
até 200 ml de água do interstício por hora sem acúmulo, ou seja, sem
edema, às custas apenas da elevação da freqüência respiratória ou da
amplitude desta. Nessa situação, o pulmão tolera aumento de pressão
hidrostática capilar pulmonar de até 35 mmHg, sem existir congestão
severa( 2, 3). Portanto, a taquipnéia pode ser a tradução da elevação
da pressão hidrostática pulmonar associada ou não à sensação subjetiva
de dispnéia, ainda sem alterações ao exame físico do paciente.
Quando a ultrafiltração excede a drenagem de líquidos, os capilares
linfáticos tornam-se completamente ingurgitados. O ultrafiltrado
excedente, inicialmente, acumula-se no interstício do pulmão nas
regiões peribrônquicas (estágio I); a seguir, ocorre acúmulo nos
septos interalveolares (estágio II); e, por último, parcialmente
(estágio IIIa) ou totalmente (estágio IIIb), na luz alveolar (Fig. 1).
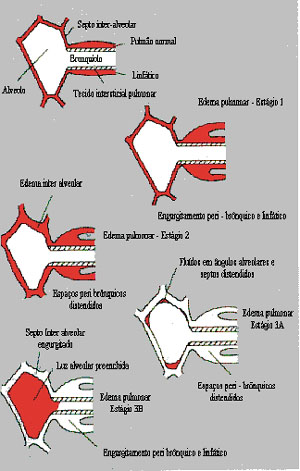 |
| Figura 1. Estágios do edema pulmonar. |
Quando esse processo se torna crônico ou a instalação do edema é
gradual, existe a adaptação dos mecanorreceptores, podendo ocorrer
edema pulmonar sem taquipnéia ou queixa de dispnéia marcante(2).
O aumento da pressão hidrostática em capilares da parede brônquica
também causa edema dessa parede, reduzindo sua luz. A mínima redução
da luz dos brônquios leva ao aumento da resistência das vias aéreas
proporcional à quarta potência da diminuição da luz, segundo a lei de
Poiseulle(2):
 |
onde: F = fluxo através do tubo, s P = gradiente de
pressão entre as extremidades do tubo, R = raio do tubo, 8 = constante
numérica igual a 8, h = viscosidade do conteúdo do tubo, P = 3,1415...
A expressão clínica do edema agudo de pulmão se dá, portanto, por um
desconforto respiratório com ou sem insuficiência resultante da soma
de uma série de fatores:
— Inundação alveolar, com conseqüente redução da complacência
pulmonar. (2, 3)
— O edema da parede dos brônquios, causado pelo aumento da pressão
hidrostática vascular nestes, reduz a luz do órgão, aumentando sua
resistência ao fluxo de ar. O edema de vias aéreas aumenta a
reatividade da musculatura brônquica, podendo agravar a obstrução
mecânica(4).
— A hipoxemia é causada pelo “shunt“ pulmonar ocasionado pelo líquido
acumulado no interstício, com conseqüente aumento da barreira
alveolocapilar, levando à maior dificuldade para a hematose. As áreas
de colapso alveolar são regiões de “shunt” verdadeiro, onde existe
passagem de sangue sem contato com a barreira alveolocapilar
funcionante. A pressão parcial de oxigênio arterial baixa é capaz de
estimular quimiorreceptores localizados na aorta e carótidas,
aumentando a sensação de dispnéia e o tônus simpático(2-4).
— A intensa atividade muscular respiratória pode elevar o fluxo
sanguíneo nessa musculatura, passando do nível normal de 4% a 5% para
até 50% do débito cardíaco, desviando o oxigênio destinado para outros
tecidos, como o do sistema nervoso central, podendo provocar variações
no nível de consciência(3).
A fadiga muscular, por causa de sua intensa atividade, leva,
progressivamente, à hipoventilação com hipoxemia, à retenção de
dióxido de carbono e à acidose respiratória, culminando com piora da
função cardíaca e da congestão. Fechase, assim, um círculo vicioso,
que, perpetuado, leva à morte(5).
ETIOLOGIA E CLASSIFICAÇÃO
Segundo a lei de Starling, o extravasamento de líquido para o terceiro
espaço pulmonar pode ocorrer por vários fatores:
— aumento da pressão hidrostática de origem cardiogênica;
— aumento da pressão hidrostática de origem não-cardiogênica, como,
por exemplo, a doença venoclusiva pulmonar;
— aumento da permeabilidade da membrana capilar, decorrente de
processos locais, como pneumonias, ou processos sistêmicos, como
pancretites, queimaduras, sepse ou síndrome de reação inflamatória
sistêmica;
— redução da pressão hidrostática intersticial, como nos rápidos e
maciços esvaziamentos de derrames pleurais;
— redução da pressão oncótica sanguínea, que, por si só, não é causa
de edema pulmonar, mas pode ser um importante fator colaborador;
— redução da drenagem linfática, como ocorre na linfangiite
carcinomatosa, silicose e doença pulmonar obstrutiva crônica, que, por
sua vez, também não é causa primária de edema pulmonar, mas, sim, um
fator colaborador.
O edema pulmonar, em geral, é classificado, temporalmente, em agudo ou
crônico, ou cardiogênico e não-cardiogênico, de acordo com o mecanismo
que o produziu(1, 2, 6-8).
O edema agudo de pulmão tem várias etiologias. As causas mais comuns
de disfunção cardiogênica aguda são a isquemia coronariana e a
emergência hipertensiva; outras mais raras são a insuficiência mitral
aguda por rotura de cordoalha tendínea (como ocorre na degeneração
mixomatosa, na endocardite e na degeneração senil) ou disfunção de
musculatura papilar e insuficiência aórtica aguda, que pode ocorrer no
trauma fechado ou na dissecção aguda de aorta. As cardiopatias
crônicas que cursam com disfunção ventricular sistólica e diastólica e
valvopatias podem causar edema pulmonar agudo quando associadas a um
fator desencadeante, em geral arritmias, infecções, isquemia,
emergência hipertensiva e uso incorreto de dieta e
medicamentos(1,6-9).
DIAGNÓSTICO
O diagnóstico de desconforto respiratório por edema agudo de pulmão
deve ser clínico e imediato para o pronto início da terapêutica. O
paciente queixa-se de dispnéia de início ou piora súbita, tendo, ao
exame físico, sinais representativos do esforço da musculatura
inspiratória, como uso dos músculos escalenos e esternocleidomastóides,
tiragem de fúrcula, intercostal e batimento de asa nasal. Associam-se,
ainda, taquipnéia e expiração forçada, com a glote semifechada, com a
intenção de conseguir pressão expiratória positiva; isso resulta no
uso da musculatura abdominal, em especial os músculos retos
abdominais, gerando, por vezes, balancins toracoabdominais. Ruídos
compatíveis com cornagem podem aparecer quando há expiração forçada. A
ausculta pulmonar é variável, podendo-se encontrar, mais comumente,
estertores crepitantes, que resultam do colapso de alvéolos e
bronquíolos terminais; porém, pode ser encontrado, apenas, murmúrio
vesicular mais rude, em decorrência de edema intersticial e
espessamento dos septos interalveolares. Roncos esparsos e sibilos são
resultantes da transudação brônquica e do edema da mucosa brônquica e
da hiper-reatividade da musculatura lisa, respectivamente. A ausculta
também pode ser normal, pois, numa primeira fase do edema agudo de
pulmão, a drenagem linfática compensa a transudação vascular,
existindo apenas a taquipnéia. A tosse pode ser manifestação de
congestão pulmonar.
Associados à dispnéia aparecem sinais de liberação adrenérgica, como
taquicardia, hipertensão, sudorese fria, palidez cutânea e ansiedade.
Outros achados podem ajudar a esclarecer a etiologia e/ou o
diagnóstico diferencial de edema pulmonar, como presença de dor
torácica compatível com insuficiência coronariana, galope cardíaco (B3
e/ou B4), sopros cardíacos e posição do “ictus cordis”, cuja
lateralização representa aumento da área cardíaca.
DIAGNÓSTICO DIFERENCIAL
Os diagnósticos diferenciais do edema agudo de pulmão são determinados
por duas de suas características: início súbito da dispnéia e presença
de congestão pulmonar clínica e radiológica.
A dispnéia súbita pode ter outras causas, como tromboembolismo
pulmonar, broncoespasmo, broncoaspiração, e inalação de gases tóxicos
e irritantes. Quanto ao acúmulo de água, no terceiro espaço pulmonar
existem outras causas, em que não há hipertensão venocapilar, como no
edema pulmonar das grandes alturas, edema neurogênico, linfangiites
pulmonares, “overdose” de narcóticos e síndrome do desconforto
respiratório agudo do adulto, com suas mais diversas etiologias.
TRATAMENTO
O tratamento do edema agudo de pulmão consiste de três etapas
sobrepostas. Na primeira etapa, o objetivo é manter as funções
respiratórias dentro de limites que permitam a manutenção da vida. Na
segunda etapa, o objetivo é a redução da pressão hidrostática capilar
pulmonar e a conseqüente redução do ultrafiltrado para o interstício
pulmonar, de forma farmacológica ou não. Por último, na terceira
etapa, o objetivo é tratar a causa ou eliminar o fator de
descompensação da cardiopatia de base(3).
O tratamento de suporte consiste, basicamente, em melhorar a
oxigenação sanguínea e reduzir o trabalho respiratório do paciente. A
oxigenação adequada do sangue tem por objetivo o transporte necessário
de O2 aos tecidos, com conseqüente metabolismo aeróbico e produção
eficaz de energia. Evita-se, dessa forma, a produção final de lactato
pela via glicolítica anaeróbica. A oxigenação pode ser implementada
pelo aumento da fração de inspiração de oxigênio por meio de cateteres
de oxigênio (fração inspiratória de oxigênio ou FiO2 máxima = 40%),
máscaras faciais (FiO2 máxima = 60%), máscaras de Venturi (FiO2 máxima
= 50% ), máscaras de alto fluxo com reservatório e válvula
unidirecional para o fluxo (FiO2 máxima = 98% a 100%) e, por último,
máscara com suporte não-invasivo de ventilação, seja com pressão
positiva contínua em vias aéreas (CPAP) ou com dois níveis de pressão,
ambos com FiO máxima = 100%(3).
2 A redução do trabalho respiratório é uma medida que evita a fadiga
da musculatura da caixa torácica e a retenção de CO2 (e,
conseqüentemente, a acidose respiratória), e reduz a ativi dade
metabólica anaeróbia da musculatura. Esta última resulta da grande
solicitação muscular somada a hipoxia hipoxêmica, causada pelo aumento
do gradiente alvéolo-arterial proporcionado pelo edema pulmonar. A
redução do trabalho inspiratório é feita com o auxílio pressórico
inspiratório aplicado por ventilador mecânico, tendo como interface
com o paciente intubação orotraqueal e máscara facial ou nasal(1, 3,
6, 10, 11).
A redução de líquido do terceiro espaço pulmonar diminui o gradiente
alvéolo-arterial, com conseqüente melhora da oxigenação. Provoca
aumento da complacência pulmonar ao reduzir o número de alvéolos
colabados, com desejável redução do trabalho respiratório. Esse
objetivo é atingido reduzindo-se a pressão hidrostática de capilar
pulmonar com posicionamento correto do paciente e uso de diuréticos,
vasodilatadores ou inotrópicos(3, 12, 13).
O paciente deve ser posicionado sentado e, sempre que possível, com os
membros inferiores pendentes, reduzindo, assim, o retorno venoso e a
pressão hidrostática capilar pulmonar. Monitorização contínua dos
batimentos cardíacos, pressão não-invasiva automática, oximetria de
pulso e obtenção de acesso venoso são medidas úteis e, portanto,
recomendadas. Oxigênio deve ser oferecido com frações inspiratórias
maiores que 60%, com o objetivo de manter a saturação periférica de
oxigênio acima de 90%(12, 13).
Existem vários esquemas terapêuticos no tratamento do edema pulmonar.
Em nossos serviços, usamos as diretrizes preconizadas pelo “Advanced
Cardiac Life Support” da American Heart Association(12). Assim, a
primeira linha de drogas usadas no tratamento do edema agudo de pulmão
é constituída por diuréticos, nitratos e morfina.
O nitrato usado mais freqüentemente é o dinitrato de isossorbida, na
dose de 5 mg, administrado por via sublingual a cada 5 minutos, desde
que a pressão arterial sistólica se mantenha acima de 90 mmHg(12, 13).
A furosemida é usada na dose de 0,5 mg/ kg até 1 mg/kg de peso por via
intravenosa. Em casos de insuficiência renal oligoanúrica, é utilizada
dose de 100 mg a 200 mg, com aplicação lenta. A resposta inicial
esperada é a melhora do desconforto respiratório, decorrente de
venodilatação; após 20 a 30 minutos ocorrerá a diurese propriamente
dita. Assim, se dentro de 20 minutos da aplicação do diurético não
obtivermos resposta diurética ou do desconforto respiratório, o dobro
da dose inicial é aplicada.
A morfina é um fármaco de grande auxílio na terapêutica do edema
pulmonar, pois: promove venodilatação, reduzindo o retorno venoso para
o coração em até 40%; diminui as eferências dos mecanorreceptores
intersticiais pulmonares, estimulados pelo aumento do fluxo e da
pressão hidrostática; e reduz a descarga adrenérgica do paciente,
pelos dois motivos previamente descritos, reduzindo, assim, a
pós-carga do coração. A dose usada é de 1 mg a 3 mg a cada 5 minutos,
monitorizando nível de consciência, freqüência respiratória, pressão
arterial, náuseas e freqüência cardíaca. A meperidina não deve ser
usada, pois tem mais efeitos colaterais, causados pelos seus
metabólitos aliados ao menor efeito hemodinâmico.
Caso o paciente permaneça desconfortável apesar dessas medidas
(pressão arterial sistólica > de 100 mmHg), são administrados
vasodilatadores venosos com infusão contínua. O nitroprussiato de
sódio, na dose de 0,1 µg/kg até 5 µg/kg por minuto, é utilizado caso o
paciente não tenha antecedente de coronariopatia, dor torácica ou
alteração isquêmica ao eletrocardiograma. Em caso de coronariopatia, a
medicação de escolha é a nitroglicerina endovenosa, na dose de 5 µg a
10 µg até 200 µg a 500 µg por minuto.
Drogas inotrópicas, como a dobutamina na dose de 2 µg/kg a 20 µg/kg
por minuto, são utilizadas quando há disfunção ventricular esquerda
associada a quadro clínico refratário, má perfusão periférica ou
hipotensão.
A hipoxemia refratária, a acidemia por acidose respiratória
progressiva, o rebaixamento do nível de consciência, bem como o
aparecimento de sinais clínicos de fadiga da musculatura respiratória
indicam a intubação orotraqueal com ventilação mecânica. Nos doentes
com infarto agudo do miocárdio e desconforto respiratório
moderado/importante, a intubação deve ser mais precoce, no sentido de
reduzir o consumo de oxigênio do coração e permitir intervenção
hemodinâmica de forma mais segura.
A ventilação mecânica aplicada com intubação traqueal aumenta a
sobrevida de pacientes com insuficiência e/ou desconforto respiratório
grave. Por outro lado, pode causar complicações, como infecção
pulmonar e barotrauma, além do aumento da permanência e da elevação
dos custos hospitalares. O tratamento da insuficiência respiratória
moderada e grave por meio da ventilação mecânica não-invasiva e da
pressão positiva contínua em vias aéreas vem ganhando espaço
recentemente. Descrita por Barach e Poulton, em 1935 e 1936,
respectivamente( 1, 6, 11, 14), a pressão positiva contínua em vias
aéreas reduz a necessidade de intubação traqueal em 30% a 35% dos
casos(1). Proporciona melhora funcional respiratória precoce em
resposta à medicação habitual(9); entretanto, não existe comprovação
da redução de mortalidade com seu uso. Apesar de suas vantagens, como
a fácil aplicabilidade, a redução do custo e as poucas complicações,
as formas não-invasivas de aplicação de pressão em vias aéreas não
isolam a via aérea, sendo, por isso, consideradas formas secundárias
de suporte.
A pressão positiva contínua em vias aéreas e a ventilação não-invasiva
podem ser aplicadas por meio de máscara nasal ou de máscara facial(1,
6-8, 11).
A pressão positiva intratorácica reduz o retorno venoso, reduzindo a
pré-carga, e a pressão transmural em parede de ventrículo esquerdo,
reduzindo a pós-carga . O apoio pressórico inspiratório reduz o
consumo de oxigênio da musculatura respiratória, que, em condições
basais, é de 5%, e passa a até 40% a 50% do débito cardíaco em
condições de estresse, reduzindo, assim, o trabalho cardíaco( 5).
Em nossos serviços, temos empregado, nos últimos anos, ventilação
não-invasiva no edema agudo pulmonar, com resultados promissores. De
1999 a 2000, 49 pacientes com edema agudo de pulmão
moderado/importante foram randomizados para tratamento em três grupos:
1) oxigenoterapia habitual com FiO2 de 50%; 2) pressão positiva
contínua em vias aéreas de 10 cmH2O e FiO2 de 50%; e 3) ventilação
não-invasiva em dois níveis de pressão, com pressão expiratória de 10
cmH2O e inspiratória de 15 cmH2O, também com FiO2 de 50%. Observamos
que os casos que utilizaram pressão positiva ao final da expiração
apresentaram redução do trabalho respiratório e cardíaco mais precoce,
além da redução da necessidade de ventilação mecânica (Fig. 2)(15).
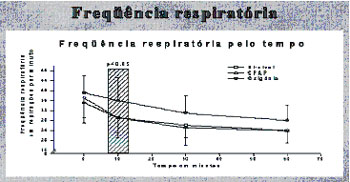 |
| A |
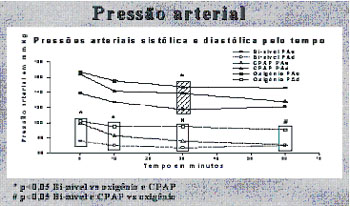 |
| B |
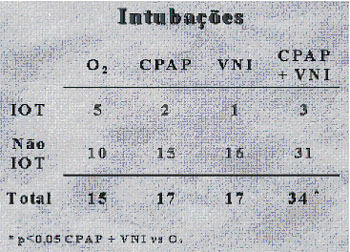 |
| C |
| Figura 2. A) Variação temporal da
freqüência repiratória. B) Variação temporal da pressão arterial.
C) Necessidade de intubação traqueal. Bi-nível = VNI, ventilação
não-invasiva em dois níveis de pressão; CPAP, pressão positiva
contínua em vias aéreas; IOT, intubação orotraqueal; O2,
oxigenoterapia. C |
REFERÊNCIAS
1. Bersten AD, Holt AW, Vedig AE, Skowronski GA, Baggoley CJ.
Treatment of severe pulmonary edema with continuous positive airway
pressure delivered by face mask. N Engl J Med 1991;325:1825-30.
2. Sharp JT, Griffith GT, Bunnell IL, Greene DG. Ventilatory mechanics
in pulmonary edema in man. J Clin Invest 1958;37:111-7.
3. Barbas CSV, Bueno MAS, Amato MBP, Hoelz C, Rodrigues-Junior M.
Interação cardiopulmonar durante a ventilação mecânica. Rev Soc
Cardiol Estado de São Paulo 1998;3:28- 41.
4. Jones JG, Lemen R, Graf PD. Changes in airway calibre following
pulmonary venous congestion. Br J Anaesthesiol 1978;50:743-51.
5. Aubier M, Trippenbach T, Roussos C. Respiratory muscle fatigue
during cardiogenic shock. J Appl Physiol 1981;51:499-508.
6. Sachetti AD, Harris RH, Paston C. Bi-level positive airway pressure
support system use in acute congestive failure: preliminary case
series. Acad Emerg Med 1995;2:714-8.
7. Lin M, Chiang HT. The efficacy of early continuous airway pressure
therapy in patients with acute cardiogenic pulmonary edema. J Formosan
Med Assoc 1991;90:736-43.
8. Lin M, Yang YF, Chiang HT, Chang MS, Chiang BN, Cheitlin MD.
Reappraisal of continuous positive airway pressure therapy in acute
cardiogenic pulmonary edema — Short term results and long term
follow-up. Chest 1995;107:1379-86.
9. Lenique F, Habis M, Lofaso F, Dubois-Randé JL, Harf A, Brochard L.
Ventilatory and hemodynamic effects of continuous positive airway
pressure in left heart failure. Am J Resp Crit Care Med
1997;155:500-5.
10. Mehta S, Jay GD, Woolard RH, Hipona RA, Conolly EM, Cimini DM, et
al. Randomized, prospective trial of bilevel versus continuous
positive airway pressure in acute pulmonary edema. Crit Care Med
1997;25:620-8.
11. Rasanen J, Heikkila J, Downs J, Nikki P, Vaissanen I, Viitanen A.
Continuous positive airway pressure by face mask in acute cardiogenic
pulmonary edema. Am J Cardiol 1985;55:296-300.
12. Hypotension/Shock/Pulmonary Edema. In: Advanced Cardiac Life
Support. 2nd ed. Dallas: American Heart Association; 1997.
p.1.40-1.47.
13. Cotter G, Metzker E, Kaluzk E. Randomized trial of high dose
isosorbide dinitrate plus low dose furosemide versus high dose
furosemide plus low dose isosorbide dinitrate in severe acute
cardiogenic pulmonary oedema. Lancet 1998;351:389-93.
14. Hoffmann B, Welte T. The use of noninvasive pressure support
ventilation for severe respiratory insufficiency due to pulmonary
oedema. Intens Care Med 1999;25(1):15-20.
15. Park M, Sangeam MC, Volpe MS, Feltrim MIZ, Nozawa E, Leite PF,
Viecili PRN, Mansur AJ, Lorenzi-Filho G. Proceedings of the XXII
Congress of the European Society of Cardiology. Aug 26-30; Amsterdam —
The Netherlands 2000.
16. Pang D, Kenan SP, Cook DJ, Sibbald WJ. The effect of positive
airway support on mortality and the need of intubation in cardiogenic
pulmonary edema — a systematic review. Chest 1998;114:1185-91.
17. Buda A, Pinsky MR, Ingels NB, Daughters GT, Stinson EB, Alderman
EL. Effect of intrathoracic pressure on left ventricular performance.
N Engl J Med 1979;301:453-9.
18. Montner PK, Greene ER, Murata GH, Stark DM, Timms M, Chick TW.
Hemodynamic effects of nasal and face mask continuous positive airway
pressure. Am J Respir Crit Care Med 1994;149:1614-8.
19. Blomqvist H, Wickerts CJ, Berg B, Frostell C, Jolin A,
Hedenstierna G. Does PEEP facilitate the resolution of extravascular
lung water after experimental hydrostatic pulmonary oedema? Eur Resp J
1991;4:1053-9.
20. Philip-Joet FF, Paganelli FF, Dutau HL, Saadjian AY. Hemodynamic
effects of bilevel nasal positive airway pressure ventilation in
patients with heart failure. Respiration 1999;66:136-43.
21. Newberry DL, Noblett KE, et al: Noninvasive bilevel positive
pressure ventilation in severe acute pulmonary edema. Am J Emerg Med
1995;13(4):479-82.
MARCELO PARK, LUIZ FRANCISCO CARDOSO