Setor de Emergências e Terapia Intensiva — Instituto Dante Pazzanese de Cardiologia Unidade de Terapia Intensiva — Hospital Municipal Dr. Arthur Ribeiro de Saboya
Endereço para correspondência:
Rua Pelotas, 323 — ap. 113 — Vila Mariana — CEP 04012-001 — São Paulo — SP
INTRODUÇÃO
Os problemas pulmonares são as causas mais significativas de morbidade no pós-operatório de cirurgia cardíaca. A insuficiência respiratória é uma complicação freqüente, e aproximadamente 5% dos pacientes requererão suporte ventilatório adicional, em decorrência de pobre oxigenação e/ou de ventilação inadequada.(1, 2)
A instalação da insuficiência respiratória encontra-se na dependência dos seguintes fatores:
1) pré-operatórios — pacientes portadores de pneumopatias, em vigência de descompensação cardíaca, ou com doenças que interfiram na função respiratória ou em seu estado nutricional;
2) intra-operatórios — serão comentados a seguir;
3) pós-operatórios — efeitos das alterações ocorridas no intra-operatório, secundárias a anestesia e circulação extracorpórea, gerando tanto alterações nas trocas na membrana alvéolo capilar pulmonar como ventilatórias. Alterações pós-operatórias concorrem ainda para: estabelecimento ou perpetuação do quadro de insuficiência respiratória; disfunção ventricular esquerda; hiper-hidratação; broncospasmo; distúrbios hidroeletrolíticos, endocrinológicos e nutricionais, gerando alterações ventilatórias; presença de efusões pleurais; mau posicionamento da cânula endotraqueal; e má programação de parâmetros ventilatórios.(1-3)
Após a circulação extracorpórea, os pulmões são mais sujeitos à disfunção que qualquer outro órgão. A acentuada diminuição ou mesmo a ausência de fluxo pulmonar durante a circulação extracorpórea resulta no decréscimo acentuado do "sheer stress" nos capilares pulmonares. Tal diminuição, associada à reação inflamatória pulmonar mediada pelo complemento, resultará em seqüestro neutrofílico na vasculatura pulmonar e peroxidação de radicais livres de oxigênio da membrana lipídica. Isso, por seu turno, produz vasoconstrição pulmonar e aumento da permeabilidade da barreira alveolocapilar pulmonar.(4)
Uma parcela da disfunção pulmonar pode ser atribuída à presença de atelectasias, cujo desenvolvimento é ligado a: graus variáveis de atelectasia residual após a reexpansão pulmonar, com o restabelecimento da ventilação após o final da circulação extracorpórea; compressão do lobo inferior esquerdo, imóvel durante a circulação extracorpórea; e lesão do nervo frênico ou disfunção diafragmática, resultando em disfunção mecânica.(4)
Durante a circulação extracorpórea, a ventilação mecânica distribui fluxo gasoso preferencialmente nas áreas não-dependentes dos pulmões. Isso causa alteração da relação ventilação/perfusão e resulta em microatelectasias nas áreas pulmonares dependentes.
Embora a depleção de surfactante pulmonar durante a circulação extracorpórea seja sugerida como fator contribuinte para a instalação de atelectasias, não existem evidências que comprovem sua ocorrência.
Após a toracotomia, a complacência dos pulmões e da caixa torácica decresce significativamente. O máximo de decréscimo ocorre em cerca de três dias, mas persiste, em menor grau, por seis ou mais dias.
Alterações na mecânica da caixa torácica levam ao decréscimo do volume expiratório forçado (VEF1) e da capacidade residual funcional (CRF). A alteração do VEF1 pode persistir por seis semanas. Ocorrerão, também, redução da força inspiratória e incoordenação na expansão da caixa torácica, culminando com decréscimo do volume corrente e da eficiência respiratória e aumento do consumo de oxigênio e do gasto energético com a função respiratória.(4)
IDENTIFICAÇÃO PRÉ-OPERATÓRIA DOS PACIENTES DE RISCO
Os pacientes com risco potencial são aqueles que apresentam os seguintes fatores:
1) Idade avançada.
2) Obesidade.
3) Tabagismo: se vigente até dois meses antes da cirurgia, associa-se com incidência quatro vezes maior de complicações pulmonares pós-operatórias; parar de fumar poucos dias antes da cirurgia aumenta a secreção mucosa pulmonar.
4) Portadores de doença pulmonar intrínseca:
a) doença pulmonar obstrutiva crônica;
b) asma ativa, particularmente dependente de corticoterapia;
c) fibrose e hipertensão pulmonar;
d) toxicidade medicamentosa (amiodarona);
e) infecção pulmonar ativa.
5) Condições gerais do paciente:
a) estado nutricional — fraqueza e ineficácia da musculatura respiratória, e maior suscetibilidade a infecções;
b) sobrecarga de volume ou insuficiência cardíaca congestiva, podendo comprometer as trocas gasosas;
c) doenças sistêmicas ativas comprometendo a competência do sistema respiratório;
d) alterações do estado mental e da função neuromuscular;
e) deformidades da caixa torácica.(1-3)
Antes da cirurgia, devem ser realizados os seguintes exames:
1) Radiografia de tórax, realizada em todos os pacientes para identificar infiltrações e massas pulmonares.
2) Testes de função pulmonar para identificar doença pulmonar obstrutiva crônica grave, caracterizada por fluxo expiratório 25-75, volume expiratório forçado no primeiro segundo e capacidade vital forçada com valores menores que 50% do previsto. A redução significativa desses parâmetros de fluxo expiratório estará associada a maior tempo de permanência na UTI e aumento da incidência de complicações pulmonares.
3) Gasometria arterial: a paCO2 elevada tem sido valorizada como marcador de morbidade e mortalidade pós-operatória. São sinais de pobre prognóstico pós-operatório:
a) paCO2 maior que 50 mmHg;
b) paO2 menor que 60 mmHg.
Mesmo nos pacientes com evidência clínica de doença pulmonar grave fica muito difícil a contra-indicação cirúrgica, pela impossibilidade da quantificação da participação da doença cardíaca nessa disfunção.(1-4)
ASSISTÊNCIA VENTILATÓRIA AO PACIENTE NO PÓS-OPERATÓRIO
Rotina de assistência ventilatória
Ao se colocar um paciente sob ventilação mecânica deve-se evitar a instalação ou perpetuação de quadro de insuficiência respiratória, observando-se dois objetivos fundamentais:
1) Ventilação com baixos níveis de pressão nas vias aéreas — os altos níveis de pressão inspiratória são responsáveis pela gênese e pela perpetuação do quadro de insuficiência respiratória. Níveis de pressão inspiratória superiores a 40 cmH2O e de pressão de platô acima de 35 cmH2O deverão ser evitados.
2) Ventilação com frações inspiradas de oxigênio (FIO2) não-tóxicas — uma FIO2 acima de 70% tem efeitos deletérios comprovados sobre os pulmões (toxicidade por oxigênio). Deve-se manter os pacientes com FIO2 igual ou inferior a 50%.(3, 5, 6)
Avaliação inicial do paciente no pós-operatório
Deverá incluir revisão do curso intra-operatório, exame físico cuidadoso, avaliação dos gases sanguíneos e radiografia simples de tórax, com o objetivo de determinar:
— posição do tubo endotraqueal (2 cm acima da carina);
— posição do cateter venoso central;
— posição da sonda nasogástrica;
— posição do cateter de artéria pulmonar;
— posição do eletrodo de marcapasso e dos fios de aço;
— posição do balão de contrapulsação intra-aórtico;
— dispositivo de circulação extracorpórea (se existente).
As complicações que podem ocorrer incluem: atelectasia, alargamento do mediastino, hemotórax ou pneumotórax, insuficiência cardíaca congestiva e hiperinsuflação pulmonar.(1, 3)
Programação do ventilador(1, 3, 5-7)
A programação do ventilador está descrita na Tabela 1.
|
|

Objetivos da ventilação mecânica
A seguir estão descritos os principais objetivos da ventilação mecânica.
1) Manter oxigenação adequada:
a) paO2 acima de 70 mmHg;
b) saturação arterial de O2 acima de 90%.
2) Manter ventilação alveolar satisfatória.
As principais alterações ventilatórias são:
a) Hipocarbia — paCO2 entre 30 mmHg e 35 mmHg com leve alcalose respiratória é aceita, e até tem efeitos benéficos. Quando intensa, com níveis de paCO2 abaixo de 30 mmHg, tem efeitos deletérios: hipocalemia e arritmias ventriculares; e deslocamento da curva de dissociação da hemoglobina para a esquerda, com conseqüente diminuição da liberação de oxigênio para os tecidos. Para corrigir a hipocarbia, é preciso diminuir a freqüência do ventilador, aumentar o espaço morto ou reduzir o volume corrente do ventilador até 8 ml/kg de peso.
b) Hipercarbia — significa ventilação inadequada, podendo ainda ser secundária a aumento da atividade metabólica (reaquecimento e tremores). Tem como principais manifestações a taquicardia, a hipotensão e as arritmias. Suas principais causas são: mau funcionamento do ventilador; mau posicionamento do tubo endotraqueal, acotovelamento ou oclusão parcial por rolha de secreção; e pneumotórax. A conduta inicial, frente à hipercarbia, inclui programação correta ou troca do ventilador, troca ou reposicionamento da cânula endotraqueal, ou drenagem torácica nos casos de pneumotórax. Nos casos de assincronia com o ventilador, a reprogramação ventilatória, com diminuição dos níveis de PEEP, a mudança do modo ventilatório para SIMV e a instituição de pressão de suporte poderão ajudar. Caso a assincronia persista, deve-se sedar o paciente e até realizar bloqueio neuromuscular, passando para assistência ventilatória total (ventilação volume-controlado). Após essas condutas, a não-correção da hipercarbia poderá significar insuficiência respiratória grave (terminal).(1, 3, 6, 7)
INSUFICIÊNCIA RESPIRATÓRIA AGUDA
Conceito e etiologia
A insuficiência respiratória é definida como a incapacidade do sistema respiratório em manter as necessidades metabólicas do organismo, resultando em hipoxia e/ou hipercarbia.
Sua etiologia está ligada a:
— fração inspirada de oxigênio baixa;
— hipoventilação;
— baixa relação ventilação-perfusão;
— "shunt" venoarterial pulmonar;
— dificuldade de difusão.
As alterações de difusão são raras, podendo ter participação na gênese da insuficiência respiratória nos casos de fibrose pulmonar e nos estados de alto débito, como na anemia intensa.(1, 3)
Classificação da insuficiência respiratória aguda
Fisiopatologia
1) Tipo I: hipoxêmica ou não-ventilatória:
— paO2 menor que 55 mmHg a 60 mmHg;
— paCO2 menor ou igual a 40 mmHg;
— G(A _ a)O2 alargado.
2) Tipo II: hipoxêmica-hipercárpnica ou ventilatória:
— paO2 menor que 55 mmHg a 60 mmHg;
— paCO2 maior que 50 mmHg;
— G(A _ a)O2 normal ou alargado.
Duração
1) Aguda — desenvolvida ao longo de minutos ou dias:
— Tipo I, associada com alcalose respiratória;
— Tipo II, associada com acidose respiratória.
2) Crônica — desenvolvida ao longo de meses ou anos:
— Tipo, associada com hipertensão pulmonar;
— Tipo II, associada com hipercarbia e alcalose metabólica(1, 3).
INSUFICIÊNCIA RESPIRATÓRIA NO PÓS-OPERATÓRIO
A insuficiência respiratória no pós-operatório pode ser aguda ou crônica (cronificada).
Insuficiência respiratória aguda no pós-operatório(1, 3)
Estará presente quando houver evidência de:
1) Oxigenação inadequada:
— paO2 menor que 60 mmHg, com FIO2 > 50%; ou
— relação paO2 /FIO2 < 150 com níveis de PEEP até 5 cmH2O e < 200 com níveis de PEEP terapêuticos (superiores a 5 cmH2O).
2) Insuficiência ventilatória:
— paCO2 > 50 mmHg durante o suporte ventilatório.
Etiologia(1, 3)
1) Problemas mecânicos: mau funcionamento do ventilador; parâmetros inadequados do ventilador (FIO2, volume corrente, freqüência respiratória); problemas com o tubo endotraqueal (mau posicionamento, acotovelamento ou oclusão); estados de baixo débito cardíaco (dessaturação venosa e "shunt" venoarterial pulmonar).
2) Problemas pulmonares: atelectasia ou colapso alveolar; edema pulmonar cardiogênico/não-cardiogênico; hemorragia intersticial; pneumonia; broncospasmo grave;microembolizações por transfusão sanguínea.
3) Problemas intrapleurais: pneumotórax; hemotórax ou efusões pleurais.
4) Problemas metabólicos: tremores, com aumento da extração periférica de oxigênio, e aumento da taxa metabólica e da produção de CO2.
5) Problemas farmacológicos: entre os fármacos que inibem a vasoconstrição pulmonar hipóxica, aumentando o "shunt" venoarterial pulmonar, destacam-se a nitroglicerina, o nitroprussiato de sódio, os bloqueadores dos canais de cálcio, e os inibidores da enzima conversora.
Manifestações(1, 3)
Entre as principais manifestações, destacam-se: taquipnéia (maior que 30 ipm) com respiração superficial; respiração paradoxal abdominal; agitação, obnubilação ou alterações do estado mental; taquicardia ou bradicardia; arritmias; e diaforese.
Conduta(1, 3, 5-7)
1) Auscultar o tórax bilateralmente, verificando-se a simetria da ventilação.
2) Auscultar o abdome, buscando identificar um deslocamento do tubo laríngeo, eventualmente posicionado no esôfago.
3) Aumentar a FIO2 do ventilador para 100%, até se identificar o fator causal. Verificar com AMBU quando houver suspeita de mau funcionamento do ventilador, o que também permitirá identificar alterações na complacência pulmonar, assim como mau posicionamento da cânula endotraqueal, intubação seletiva e vazamentos. Nos casos em que houver dúvida quanto à cânula endotraqueal, esta deverá ser trocada rapidamente, antes que as condições hemodinâmicas se deteriorem.
4) Verificar o funcionamento e os parâmetros do ventilador. Otimizar sua programação quanto a sensibilidade, PEEP, "peak flow" e freqüência respiratória. O aumento agudo do pico de pressão inspiratória poderá significar: desenvolvimento de pneumotórax; broncospasmo grave; edema pulmonar; oclusão de cânula endotraqueal.
5) A drenagem torácica será imperiosa, quando se identificar: pneumotórax em pacientes sob ventilação mecânica, e efusões pleurais ou hemotórax de grande monta.
6) Avaliar e otimizar os parâmetros hemodinâmicos, pois o baixo débito cardíaco diminui o transporte periférico de oxigênio, diminui a saturação venosa mista, e aumenta o "shunt" venoarterial pulmonar. Quando houver necessidade do uso de medicamentos inotrópicos positivos, e os níveis tensionais permitirem, deve-se dar preferência àqueles sem efeito vasoconstritor periférico, como a dobutamina, para aumentar o transporte periférico de oxigênio (DO2).
A anemia, caso presente, deve ser corrigida, com o objetivo de manter a hemoglobina sérica em torno de 12 g% e o hematócrito, em aproximadamente 34%. Os níveis de DO2 e o consumo de oxigênio (VO2) devem ser otimizados, no sentido de reverter ou evitar a ocorrência de acidose láctica.
7) Adicionar PEEP ao circuito do ventilador, buscando a manutenção da paO2 acima de 70 mmHg e da saturação de oxigênio acima de 90%, com ventilação com níveis não-tóxicos de oxigênio, preferencialmente com FIO2 inferior a 50%.
8) Deve-se evitar níveis elevados de pressão de pico inspiratório (acima de 40 cmH2O) e pressão de platô (acima de 35 cmH2O), com a diminuição do volume corrente a níveis inferiores a 7 ml/kg, com o objetivo de manter a pressão de platô < 35 cmH2O. Quando a complacência pulmonar se encontra acentuadamente reduzida, recomenda-se ventilação com baixo volume corrente, igual ou mesmo inferior a 5 ml/kg de peso. Compensa-se parcialmente o volume minuto por meio do aumento da freqüência respiratória, ou permite-se a ocorrência de hipercapnia. Finalmente, pode-se usar a ventilação com pressão controlada.
9) A sedação e a paralisia muscular geralmente melhoram a eficácia da ventilação, reduzindo seu gasto energético por meio do relaxamento do diafragma e da parede torácica.
10) Outras medidas de suporte:
— Diuréticos (furosemida) — geralmente melhoram a oxigenação nas fases precoces de pós-operatório, quando o edema intersticial pode comprometer as trocas gasosas.
— Broncodilatadores (aminofilina e beta-estimulantes) e corticoterarapia nos pacientes com broncospasmo.
— Antibioticoterapia na suspeita de infecção.
Insuficiência respiratória crônica (cronificada) no pós-operatório(1, 3)
A insuficiência respiratória crônica caracteriza-se pela incapacidade em se retirar o paciente do ventilador nas primeiras 48 a 72 horas após a cirurgia, por comprometimento primário da oxigenação ou por insuficiência ventilatória. A forma extrema do desenvolvimento da insuficiência respiratória crônica é a síndrome da angústia respiratória aguda (SARA).
Insuficiência respiratória do tipo I ou
hipoxêmica(1, 3)
Geralmente é conseqüência de problemas preexistentes, como insuficiência cardíaca com edema pulmonar ou doença parenquimatosa pulmonar, como doença pulmonar obstrutiva crônica e hipertensão pulmonar.
As causas primárias de insuficiência respiratória crônica são:
1) Instabilidade hemodinâmica — particularmente estado de baixo débito cardíaco que requeira tratamento vasopressor.
2) Doenças pulmonares parenquimatosas:
— edema intersticial pulmonar — SARA;
— pneumonia;
— obstrução das vias aéreas inferiores — geralmente associada a doença pulmonar obstrutiva crônica.
Insuficiência respiratória tipo II ou insuficiência ventilatória(1, 3)
É a insuficiência ventilatória primária resultante do desequilíbrio entre a capacidade ventilatória e a demanda de oxigênio. É causa mais comum de insucesso de desmame no pós-operatório, por aumento do trabalho respiratório.
Etiologia(1, 3)
1) Aumento da produção de CO2 e da demanda ventilatória: febre, sépsis, tremores; dor e ansiedade; estados hipercatabólicos; oferta excessiva de carboidratos.
2) Decréscimo do "drive" respiratório: estado mental alterado; lesão neurológica ou encefalopatia.
3) Decréscimo da função dos músculos respiratórios: desnutrição protéica com fraqueza muscular; anormalidades metabólicas (hipofosfatemia, hiper ou hipomagnesemia, hipocalemia ou hipocalcemia); paralisia diafragmática ou lesão de nervo frênico.
As manifestações clínicas são similares às da insuficiência respiratória aguda.
Conduta(1, 3)
1) Melhorar o estado hemodinâmico.
2) Tratamento da hipoxemia e diminuição da impedância do ventilador: uso de PEEP; aspiração, fisioterapia respiratória, mobilização; broncodilatadores; transfusão sanguínea; diuréticos na sobrecarga hídrica; aumento do calibre da cânula endotraqueal.
3) Reduzir os requerimentos da ventilação minuto: analgesia para a dor e sedativos para a ansiedade; tratamento de infecções — pneumonia e sépsis (reduzir a febre com antitérmicos; antibioticoterapia profilática; descontaminação seletiva do tubo digestivo, discutível pelo seu alto custo — apesar de reduzir a incidência de pneumonia, não reduz a mortalidade global; sucralfate para proteção gástrica — os inibidores H2, como a cimetidina, aumentam o pH gástrico, com aumento da incidência de pneumonia nosocomial).
4) Melhorar o "drive" respiratório e a fraqueza muscular: proporcionar adequado aporte nutricional para melhorar a força muscular respiratória e a capacidade imunológica; evitar dietas ricas em carboidratos e com excesso de proteínas, dando preferência às dietas ricas em lípides; selecionar um modo adequado de suporte ventilatório para reduzir o trabalho respiratório e manter a atividade da musculatura respiratória (sempre que possível, deve-se dar preferência à associação de SIMV com pressão de suporte).
5) Fisioterapia respiratória.
6) Otimizar o equilíbrio ácido-básico, hidroeletrolítico e endócrino; a alcalose metabólica e o hipotireoidismo inibem o "drive" respiratório normal.
7) Avaliar a mobilidade diafragmática — radioscopia ou fluoroscopia.
DOENÇAS ESPECÍFICAS
SARA — síndrome da angústia respiratória aguda
Fisiopatologia(7, 8)
Após instalação do fator predisponente, circulação extracorpórea prolongada ou por reação à administração de produtos sanguíneos ocorrerá a liberação dos fatores mediadores da inflamação e a conseqüente alteração da permeabilidade da membrana alveolocapilar.
Ocorre, precocemente, grande afluxo de neutrófilos para o território pulmonar e uma série de reações:
a) Liberação de proteases (colagenase e elastase) relacionadas diretamente com a agressão à membrana celular.
b) Liberação de radicais superóxidos pelos polimorfonucleares, que alteram a estrutura secundária de proteínas e lípides da membrana celular, levando à lesão tecidual.
c) A cicloxigenase e a lipoxigenase (liberadas pelos próprios neutrófilos) na presença de ácidos graxos livres resultantes da lesão das membranas produzem prostaglandinas e leucotrienos. Estes geram vasoconstrição e broncoconstrição. Os leucotrienos possuem também efeito quimiotático para neutrófilos (LTB-4) e alteram a permeabilidade da membrana alveolocapilar.
d) Os neutrófilos recrutados ao parênquima pulmonar são ativados e agregam-se à membrana do endotélio. Essa interação entre neutrófilo e membrana endotelial é fundamental para criar um ambiente em que radicais superóxidos e proteases, liberados pelo neutrófilo ativado, possam agir sem sofrer mediação por agentes normalmente presentes na corrente sanguínea. Essa interação é feita por moléculas de adesão, as selectinas, que são expressas na membrana, tanto no próprio neutrófilo (L-selectina) como na célula endotelial (E-selectina e P-selectina). A expressão das moléculas de adesão ocorre em resposta a um estímulo por citoquinas (como, por exemplo, fator de necrose tecidual), e facilita a agregação dos polimorfonucleares. Essa interação é feita por moléculas de adesão, as selectinas, que são expressas na membrana tanto do próprio neutrófilo (L-selectina) como na célula endotelial (E-selectina, P-selectina), em resposta ao estímulo por citoquinas (como, por exemplo, fator de necrose tumoral) ou mediadores da inflamação (como lipopolissacárides — LPS). Além das selectinas, a família das integrinas tem papel importante nesse mecanismo de adesão dos neutrófilos. Uma vez ativadas por mediadores como o TNF, passam a expressar receptores de superfície CD11/CD18, que reconhecem as moléculas de adesão intercelulares ICAM-1 e ICAM-2 expressas na superfície do endotélio. Essa interação protéica torna ainda mais forte a adesão, permitindo que a reação inflamatória prossiga localmente.
e) Os neutrófilos ativados liberam citoquinas como o fator de necrose tumoral e as interleucinas, principalmente IL-1, IL-4, IL-6 e IL-8, que têm papel determinante no desenvolvimento da SARA. Em resposta a esses mediadores, verifica-se aumento da atividade pró-coagulante, com deposição de fibrina, aumento da degradação e liberação de produtos de degradação da fibrina e dímero D. Esses fenômenos ocorrem em toda a área lesada, mas principalmente no lado alveolar da barreira, contribuindo para a formação da membrana hialina, que é a marca histológica da síndrome.
f) A presença de fibrina contribui para a inativação do surfactante. Serve de matriz e estímulo para a proliferação e a ativação de fibroblastos, responsáveis pela fibrose e pela reparação do tecido alveolar. Produtos de degradação da fibrina podem aumentar a permeabilidade vascular e a vasoconstrição, uma associação que favorece a formação de edema. Níveis elevados de dímero D e de atividade pró-coagulante no lavado broncoalveolar de pacientes de risco são marcadores do desenvolvimento da síndrome.
A ocorrência da síndrome em indivíduos depletados de neutrófilos mostra que, além do neutrófilo, dois efeitos têm papel destacado em seu desenvolvimento: os macrófagos e as plaquetas.
Os macrófagos, células residentes no território pulmonar, contam com um arsenal lesivo semelhante ao dos neutrófilos e produzem grande quantidade de fator de necrose tumoral. Essa citoquina é capaz de desencadear todo o processo da SARA.
As plaquetas, por sua vez, por meio de sua agregação intravascular e conseqüente formação de microtrombos, levam ao desequilíbrio na relação V/Q, que piora pela vasoconstrição secundária à liberação de prostaglandinas e serotonina. As plaquetas também estão envolvidas no processo de reparação que se segue à lesão do parênquima pulmonar, já que liberam grandes quantidades de PGDF, potente estimulante mitogênico para os fibroblastos.
Recentemente demonstrou-se a importância de um novo mediador da inflamação: o óxido nítrico. Liberado por endotélio, neutrófilos, macrófagos e plaquetas, participa diretamente da alteração da permeabilidade da membrana alveolocapilar.
Numa fase mais avançada, há uma certa organização do processo, com proliferação de fibroblastos e deposição de colágeno, culminando com o estabelecimento de fibrose. A fibrose pulmonar depende não somente da presença de mitógenos, como o PGDF, como também de uma matriz apropriada, que resulta da coagulação intra-alveolar da fibrina extravasada do plasma, em função da alteração da permeabilidade da membrana alveolocapilar.
A superfície epitelial dos alvéolos apresenta intensa atividade anticoagulante determinada pela presença de ativadores do plasminogênio. A perda desses ativadores é um dos eventos iniciais na SARA.
A surfactante sofre interferência em sua ação pelos produtos de degradação da fibrina e tem sua produção alterada, por causa do comprometimento dos pneumócitos do tipo II.
Essas alterações causam anormalidades na função pulmonar. Há diminuição da complacência pulmonar e estabelecimento de insuficiência respiratória e hipoxêmica secundária a alterações na relação ventilação-perfusão, e ao estabelecimento de "shunt" venoarterial pulmonar secundário ao colapso alveolar. O broncospasmo, se presente, contribuirá para a intensificação dessas alterações.(7, 8)
Correlação anátomo-fisiopatológica-clínica (9)
1) Fase aguda
a) Fase latente
— Patologia: desenvolvimento de lesão da membrana alveolocapilar.
— Fisiopatologia: pouca evidência de comprometimento pulmonar e aumento do fluxo linfático.
— Clínica: a alteração etiopatogênica primária domina o quadro.
b) Fase de edema intersticial
— Patologia: lesão extensa na membrana alveolocapilar; lesão do septo interalveolar; grande edema intersticial rico em proteínas.
— Fisiopatologia: fluxo linfático acentuadamente aumentado, pulmão rígido (complacência pulmonar acentuadamente diminuída), alteração da relação ventilação-perfusão.
— Clínica: apreensão e dispnéia, radiografia de tórax (normal ou imagens intersticiais), gases (alcalose respiratória + hipoxemia + G(A-a)O2 elevado).
c) Fase de edema intra-alveolar
— Patologia: pulmões úmidos e pesados, líquido intra-alveolar pulmonar rico em proteínas, anormalidade na capacidade funcional da surfactante.
— Fisiopatologia: capacidade residual funcional e complacência pulmonar acentuadamente diminuídas, aumento das alterações na relação ventilação-perfusão, "shunt" venoarterial pulmonar significativo.
— Clínica: taquipnéia acentuada, estertores difusos à ausculta pulmonar (radiografia de tórax: pulmão branco, fino infiltrado alveolar difuso), gases (hipoxemia intensa, alcalose respiratória, G(A-a)O2 acentuadamente aumentado).
2) Fase crônica
— Patologia: membranas hialinas intra-alveolares, proliferação de pneumócitos tipo II, fibrinogênese (destruição da membrana alveolocapilar).
— Fisiopatologia: intensa alteração da relação ventilação-perfusão, complacência pulmonar acentuadamente diminuída.
— Clínica: pressão de pico inspiratório com aumento gradativo, radiografia de tórax = fibrose pulmonar, gases (piora progressiva do G(A-a)O2, presença ou não de hipercarbia), morte (falência respiratória e/ou causa associada).(9)
Diagnóstico(3, 7, 9, 10)
A suspeita clínica inicial é caracterizada pela incapacidade em se manter uma saturação e uma pressão parcial de oxigênio no sangue arterial em níveis aceitáveis, com acréscimos sucessivos na fração inspirada de oxigênio (FIO2).
A caracterização do quadro ocorre com o aparecimento de um padrão radiológico característico: padrão alveolar difuso, que será de ocorrência mais tardia; de uma relação paO2 /FIO2 inferior a 150, nos pacientes com PEEP < 5 cmH2O; e inferior a 200, se o PEEP for superior a 5 cmH2O.(3, 7, 9, 10)
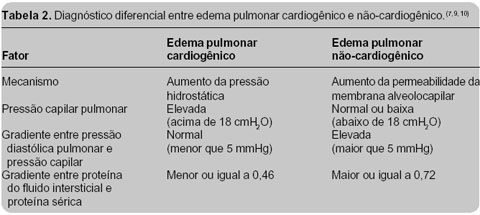
A monitorização hemodinâmica com cateter de Swan-Ganz visa ao diagnóstico diferencial entre o edema pulmonar não-cardiogênico (não-hidrostático) e o edema pulmonar cardiogênico (hidrostático). A SARA cursa com pressão capilar ("wedge") normal ou muito próxima do normal, ao passo que no edema pulmonar cardiogênico ocorre pressão capilar elevada, geralmente acima de 20 mmHg.
A Tabela 2 apresenta, de forma resumida, o diagnóstico
diferencial entre edema pulmonar cardiogênico e não-cardiogênico.
|
|
Tratamento(1, 3, 7, 9, 10)
1) Antibioticoterapia — caso não haja culturas para direcioná-la, dá-se preferência a uma cefalosporina de terceira geração associada a um aminoglicosídeo.
2) Manutenção de equilíbrio hídrico adequado, com a administração de diuréticos (furosemida) se houver sinais de hiper-hidratação. Em caso de necessidade de expansão volêmica, esta poderá ser feita tanto com colóide como com cristalóide. Não há dados conclusivos, na literatura, que demonstrem alguma superioridade do uso de colóides sobre cristalóides.
3) Otimização do DO2 em níveis supranormais, visando à oferta periférica de oxigênio adequada e à reversão tanto da hipoxia tissular como da acidose láctica, se existente. Durante a SARA, ocorre a diminuição da taxa de extração de oxigênio. É importante ressaltar que não há um valor supranormal fixo de DO2 e VO2, os quais devem ser mantidos em valores que propiciem a reversão do estado de hipoxia tissular, espelhada por marcadores como lactato sérico. Tais objetivos podem ser alcançados por meio de:
— manutenção de um estado normovolêmico;
— transfusão sanguínea, buscando a manutenção da hemoglobina sérica em torno de 12 g% e do hematócrito em torno de 34%; e
— uso de medicamentos inotrópicos positivos, preferencialmente sem efeito vasoconstritor periférico, como a dobutamina (efeito beta1 estimulante).
O quadro hemodinâmico inicial da SARA caracteriza-se por débito cardíaco elevado e baixa resistência vascular sistêmica. Somente nos casos em que a administração de dobutamina associada à expansão volêmica não for suficiente para a manutenção de pressão de perfusão efetiva para a perfusão de órgãos nobres (cérebro e coração) é que se justifica o uso de medicamentos com ação vasoconstritora (alfa-estimulantes).
4) Ventilação mecânica — o melhor modo ventilatório é aquele capaz de gerar pressão acima da pressão de abertura dos alvéolos durante curto espaço de tempo, trabalhando a maior parte do ciclo respiratório com pressões pouco acima da pressão crítica de colabamento alveolar.
a) Para se evitar barotrauma:
— volume corrente de 8-6 ml/kg de peso, sempre que possível;
— tentar manter pico de pressão inspiratória abaixo de 40 cmH2O; e pressão de platô abaixo de 35 cmH2O;
— evitar assincronia entre o paciente e o ventilador;
— evitar AMBU;
— utilizar sempre fluxo decrescente ou pressão controlada.(1, 3, 7, 9, 10)
b) Controle da hipoxemia:
— Cálculo do PEEP pelo método
melhor complacência î
8 a 20 cmH2O
curva pressão-volume ì
Sempre que forem utilizados altos níveis de PEEP, a monitorização hemodinâmica torna-se necessária.
— Inversão da relação inspiratória/expiratória: 2:1 a 4:1 — pressão controlada (ciclada a tempo e limitada à pressão).
— Casos refratários: ventilação com pressão positiva e baixa freqüência + oxigenação por membrana extracorpórea.
c) Com essas medidas, objetiva-se:
— utilizar FIO2 inferior a 70%, preferencialmente < 50%;
— manter os níveis de pressão inspiratória de pico < 40 cmH2O, e pressão de platô < 35 cmH2O;
— aumentar a capacidade residual funcional dos pulmões;
— recrutamento alveolar.
5) Modos de ventilação mecânica na SARA:
a) Ventilação com volume controlado (+ PEEP)
Esse é o modo ventilatório que precisa ser tentado inicialmente. O ventilador deve ser programado da seguinte forma:
— Modo: assistido-controlado.
— Sensibilidade: mínima (geralmente 0,5 cmH2O).
— Volume corrente: alterado de acordo com as alterações da complacência pulmonar. Inicia-se com volume corrente de 8 ml/kg de peso, podendo ser reduzido até níveis de 6 ml/kg de peso, com base nos valores da pressão de pico inspiratório, desde que em vigência de PEEP.
— Freqüência respiratória: entre 8 ipm e 14 ipm. Em geral, é mantida em freqüências mais elevadas, para compensar parcialmente o volume minuto, na presença de volume corrente reduzido.
— PEEP: é introduzido com o intuito de ventilar o paciente com as menores frações inspiradas de oxigênio, mantendo oxigenação arterial satisfatória. Seu objetivo básico é o aumento da capacidade residual funcional. O PEEP pode ser calculado por dois métodos:
A) Curva de pressão x volume = PEEP ideal(7, 11, 12)
(Fig. 1).
|
|
|
Figura 1. Curva P x V estática do sistema respiratório.(6) |
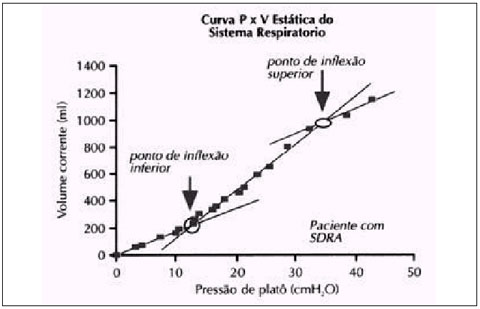
Nesse gráfico, o primeiro ponto de inflexão (pflex1) representa a pressão crítica de colabamento alveolar. Uma pressão acima desta recrutará a maioria dos alvéolos — valores de 8-20 cmH2O.
B) Método da melhor complacência = PEEP mínimo(7, 13, 14). É um nível de PEEP igual ou inferior a 15 cmH2O, obtido com o paciente ventilado a um volume corrente igual ou inferior a 8 ml/kg de peso. São calculadas, sucessivamente, as complacências pulmonares nos diferentes níveis de PEEP, isto é, em 15 cmH2O, posteriormente em 14 cmH2O, depois em 13 cmH2O, e assim sucessivamente até o valor de 8 cmH2O. O nível de PEEP em que se obtiver a melhor complacência pulmonar será o utilizado. A fórmula utilizada é a seguinte:
complacência pulmonar = volume corrente pressão de platô - PEEP
b) Ventilação com pressão controlada + relação inspiratória/expiratória invertida + PEEP
É utilizado quando os resultados são insatisfatórios com o modo empregado anteriormente. São mantidos os níveis de PEEP calculados pelo método da melhor complacência. O ventilador deve ser programado da seguinte forma:
— Ciclar o ventilador a tempo.
— Programar o nível de pressão desejado (sempre inferior a 40 cmH2O), suficiente para manter volume corrente adequado.
— Programar a FIO2 e a freqüência desejadas.
— Programar o tempo inspiratório: por exemplo, para uma freqüência de 20 ipm, com relação I:E de 2:1, FR = 20 ipm, então são 20 incursões/60 segundos. Cada ciclo inspiratório, portanto, terá 3 segundos. Logo, para uma relação I:E de 2:1, dois terços de 3 segundos = 2 segundos = tempo inspiratório (programado no ventilador); um terço de 3 segundos = 1 segundo = tempo expiratório.
O ventilador possibilita a programação do tempo inspiratório e da freqüência respiratória. A relação I:E poderá variar de 2:1 a 4:1. Esse modo ventilatório impõe um mínimo de resistência do paciente ao ventilador, visto que, ao se programar uma pressão fixa a ser atingida nas vias aéreas, o volume corrente será inversamente proporcional à resistência imposta ao fluxo aéreo. Estará também na dependência da resistência das vias aéreas e da complacência pulmonar. Os pacientes, portanto, deverão ser sedados e, se necessário, submetidos a curarização.
Com esses cuidados, o índice de mortalidade da SARA situa-se em torno de 50%, quando não ocorre comprometimento de outros órgãos.(3)
6) Comentários finais
a) Posição prona — o colapso alveolar na SARA guarda relação com as forças gravitacionais. Assim, a posição prona determina descompressão e reexpansão alveolar dos segmentos dorsais, áreas de maior atelectasia e edema durante o tratamento convencional em posição supina. Essa manobra não afeta a distribuição da perfusão pulmonar. Por meio de tomografia computadorizada do tórax, verifica-se que a mudança para a posição prona gera deslocamento das densidades paravertebrais para a região anterior. Na posição retroesternal, portanto, forma-se novo edema, enquanto se reabsorve o edema previamente existente na região dorsal. A região dorsal possui maior volume disponível para a ventilação, visto que o coração ocupa espaço relativamente significativo da região anterior da caixa torácica. Em alguns casos não se observa resposta à posição prona, o que pode estar relacionado com a formação de novo edema na região ventral. Os pacientes que respondem ao prona terão esse efeito aparente no período de três horas, o que dispensa períodos prolongados de prona.(15, 16) O uso dessa manobra no pós-operatório de cirurgia cardíaca encontra dificuldades em decorrência da cicatriz cirúrgica e da presença de drenos pleurais e mediastinais. Nossa experiência, portanto, é bastante pequena.
b) Óxido nítrico — o óxido nítrico administrado sob via inalatória em baixas concentrações é um vasodilatador pulmonar com meia-vida de alguns segundos. Quando na circulação, é inativado rapidamente ao se ligar à hemoglobina. Entre suas principais vantagens, destacam-se:
— Produz vasodilatação exclusivamente pulmonar, sem qualquer vasodilatação periférica, freqüentemente encontrada com os demais vasodilatadores pulmonares.
— Ao contrário dos outros vasodilatadores pulmonares que pioram o equilíbrio V/Q, o óxido nítrico, por vasodilatar somente as áreas do pulmão que estão sendo ventiladas, melhora a relação V/Q.
Estudos multicêntricos recentes têm demonstrado que o óxido nítrico inalatório, um vasodilatador pulmonar seletivo, diminui as pressões na artéria pulmonar em pacientes com hipertensão arterial pulmonar associada a SARA, porém não altera o curso e a mortalidade da síndrome.(7)
c) Corticosteróides — quando usados em altas dose e precocemente, teoricamente, teriam os seguintes efeitos benéficos:
— inibir a agregação dos granulócitos induzida pelo complemento;
— prevenir o aumento da permeabilidade vascular induzida por endotoxina;
— dificultar a aderência dos polimorfonucleares ao endotélio da microcirculação;
— inibir a fosfolipase e diminuir a síntese de tromboxano A2 e de outros metabólitos do ácido araquidônico;
— inibir a produção de radicais tóxicos de oxigênio.
Estudos multicêntricos demonstraram que os corticosteróides não previnem o desenvolvimento da SARA, e que seu uso em altas doses pode provocar elevação da mortalidade por aumento da taxa de infecção. O uso de altas doses de corticosteróides na SARA, portanto, não está justificado.(7) O uso de baixas doses de corticosteróides nas fase fibroproliferativa da SARA (uma semana de história), particularmente a metilprednisolona, na dosagem de 2 mg/kg/dia, poderia levar à melhora da função pulmonar e da sobrevida desses pacientes, por diminuir o grau da fibrose intersticial pulmonar.(7)
Broncospasmo
O desenvolvimento de broncospasmo grave pode gerar dificuldades na ventilação mecânica, como problemas hemodinâmicos, às vezes até mimetizando tamponamento cardíaco.
O broncospasmo pode ser precipitado por sobrecarga hídrica, reação a medicamentos, transfusão sanguínea, e uso de betabloqueadores. Poderá ocorrer em pacientes sem antecedente de doença pulmonar obstrutiva crônica ou fenômenos broncospásticos prévios.(3)
Tratamento
1) Broncodilatadores: inalação com soro fisiológico (0,9%, 10 ml, 4 a 6 vezes por dia) ou fenoterol (5 a 10 gotas).
2) Beta-estimulantes por via subcutânea (excepcionalmente): terbultalina (0,125 mg, 4 vezes por dia).
3) Aminofilina
— Efeitos benéficos: broncodilatador, diurético leve, aumenta o "drive" respiratório, melhora a função muscular respiratória, e pode decrescer a pressão de artéria pulmonar e melhorar a função ventricular direita.
— Efeitos deletérios: arritmogênico, efeito cronotrópico positivo, pode aumentar o "shunt" venoarterial pulmonar.
— Dosagem:
a) Ataque — 5 8 mg/kg de peso em 30 minutos, por infusão contínua.
b) Manutencão: não fumantes, 0,6 mg/kg/hora; fumantes, 0,9 mg/kg/hora; descompensação cardíaca ou doença hepática, 0,3 mg/kg/min.
4) Corticoterapia
Utilizada quando as medidas anteriores não surtirem efeito. Os corticosteróides aumentam a responsividade das vias aéreas aos beta-estimulantes.
— Dosagem: 200 mg de hidrocortisona, por via intravenosa, a cada 6 horas.(3)
Doença pulmonar obstrutiva crônica
A seguir encontra-se o padrão preconizado de assistência ventilatória:
— Cânula endotraqueal com diâmetro maior ou igual a 8 cm, principalmente se houver broncospasmo.
— Sensibilidade máxima do ventilador.
— Pressão de suporte em torno de 25 cmH2O.
— Nos pacientes com hipercapnia prévia, deve-se manter a paCO2 entre 45 mmHg e 65 mmHg, com pH dentro dos limites da normalidade.
— Medidas para diminuir a produção de CO2.
— Evitar o aporte excessivo de carboidratos, controle de infecção e da hipertermia.
— Fluxo inspiratório decrescente.
— Baixa freqüência respiratória (menor que 12 ipm).
— Altos fluxos inspiratórios (maior que 50 litros por minuto).
— Aumentar o tempo expiratório para facilitar a eliminação de CO2 (3, 5).
Dificuldade de desmame por debilidade da musculatura respiratória
Por vezes, não se consegue retirar o paciente do ventilador ou ele precisa ser reintubado em decorrência de sinais de fraqueza e fadiga da musculatura respiratória, com manutenção de volume corrente inadequado, resultando em taquipnéia compensatória. Ocorre com maior freqüência em pacientes com estado nutricional comprometido, particularmente naqueles com caquexia cardíaca, e com níveis acentuadamente elevados de hipertensão arterial pulmonar.
Durante a assistência ventilatória, poderão ser exigidos níveis mais elevados de pressão de suporte, no sentido de se manter volume corrente e freqüência respiratória adequados. A pressão de suporte será reduzida gradual e lentamente, às vezes com redução de 2 cmH2O a cada 24 horas. Com pressão de suporte de 14 cmH2O a 15 cmH2O, deve-se alternar períodos de CPAP de 5 cmH2O, inicialmente por 15 minutos a cada hora, aumentando-se gradativamente até uma hora, com o cuidado de se manter a ventilação com pressão de suporte durante o período noturno, para evitar fadiga da musculatura respiratória.
Quando o paciente estiver respirando em CPAP de 5 cmH2O (no esquema de alternância), por uma hora, inicia-se novamente a redução gradativa da pressão de suporte até atingir pressão de suporte de 8 cmH2O. Nesse ponto, caso as condições ventilatórias permaneçam estáveis, pode-se efetuar a extubação.(1, 3)
Lesão de nervo frênico e paralisia diafragmática
Etiologia
— Lesão de nervo frênico próximo ao pericárdio pelo uso de solução salina gelada — lesão por resfriamento.
— Lesão direta de nervo frênico ou devascularização durante a dissecção da artéria mamária interna esquerda.
— Lesão cirúrgica direta do nervo frênico durante a realização da incisão em "V" no pericárdio, para permitir melhor relevo do pedículo da artéria mamária interna.
Apresentação
— Lesão unilateral — muitos pacientes são oligossintomáticos, embora aqueles com doença pulmonar associada possam ter dificuldade ventilatória (respiração superficial).
— Lesão bilateral — produz, geralmente, taquipnéia, respiração abdominal bilateral e retenção de CO2 durante a tentativa de desmame do respirador.
Avaliação
— Radiografia de tórax — elevação da hemicúpula diafragmática durante a respiração espontânea. Não pode ser notada durante a ventilação mecânica.
— Fluoroscopia diafragmática — demonstra a movimentação inferior do diafragma, durante a respiração espontânea, se a paralisia for unilateral.
Tratamento
— Tratamento de suporte até a recuperação da função do nervo frênico, que pode levar até dois anos.
— Plicatura diafragmática — proporciona melhora sintomática e objetiva nos pacientes com dispnéia intensa por paralisia frênica unilateral.
— Suporte ventilatório mecânico prolongado — em geral é necessário para os pacientes com comprometimento bilateral. Alguns pacientes poderão receber assistência ventilatória domiciliar.(1, 3)
Pneumotórax
Pode ocorrer pequena abertura pleural durante a cirurgia, não sendo percebida e drenada, evidenciando-se, posteriormente, na radiografia de tórax.
A drenagem torácica deve ser efetuada independentemente da extensão do pneumotórax se o paciente estiver sob ventilação mecânica.
A possibilidade de pneumotórax hipertensivo deve sempre ser cogitada quando ocorrerem alterações nos gases sanguíneos ou instabilidade hemodinâmica sem motivo aparente. O primeiro sinal será o aumento do pico de pressão inspiratória.
A realização da radiografia de tórax, após a retirada do dreno pleural, é imperiosa, pois poderá ocorrer pequeno pneumotórax. Se a área for inferior a 20%, poderá ser observado sem a necessidade de drenagem imediata.
REFERÊNCIAS
1. Bojar MR. Respiratory management. In: Manual of Perioporative Care in Cardiac Surgery. 3ed. Boston: Blackwell Scientific Publications; 1999. p.177-212.
2. Baumgarthner WA, Owens SG, Camerom DE, Bertz BA. The Johns Hopkins Manual of Cardiac Surgical Care. St Louis: Mosby; 1994. p.161-82.
3. Timerman A, Sousa JEMR, Piegas LS, Bianco ACM. Pós-operatório de cirurgia cardíaca — insuficiência respiratória. In: Urgências Cardiovasculares. São Paulo: Sarvier; 1996. p.341-72.
4. Morris MD, St Clair Jr D. Management of patients after cardiac surgery. Curr Probl Cardiol 1999;167-227.
5. Knobel E, Meyer EC, Barbas CSV, Lorenzi Filho G. Insuficiência respiratória aguda. In: Condutas no Paciente Grave. 2ed. São Paulo: Atheneu; 1999. p.281-95.
6. Knobel E, Meyer EC, Barbas CSV, Amato MBP, Rodrigues Jr M. Ventilação artificial aplicada. In: Condutas no Paciente Grave. 2ed. São Paulo: Atheneu; 1999. p.353-79.
7. Barbas CSV, Amato MBP. Síndrome do Desconforto Respiratório Agudo. Malorca: Permanyer Publications; 1998. 89p.
8. Pittet JF, Mackersie RC, Martin TR, et al. Biological marker of acute lung injury: prognosis and pathogenetic significance. Am J Resp Crit Care 1997;155:1187-205.
9. Rippe MD, Irwin RS, Alpert JS, Dalen JE, Pratter MR. Respiratory failure V: adult respiratory distress syndrome. In: Intensive Care Medicine. Boston: Little Brown; 1985. p.404-12.
10. Petty TL, Ashbaugh DG. The adult respiratory distress syndrome: clinical features factors influencing prognosis and principles of management. Chest 1971;60:233-9.
11. Amato MBP, Barbas CSV, Meyer EC, et al. Limitations of the P-V curve in detecting alveolar hyperinflation during mechanical ventilation in ARDS. Am J Resp Crit Care Med 1995a;151:A432.
12. Gattinoni L, Pesenti A, Avali R, et al. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am Rev Respir Dis 1987; 136:730-6.
13. Gonçalves JL. Pressão positiva expiratória final. In: Terapia Intensiva Respiratória — Ventilação Artificial. Curitiba: Editora Lovise; 1991. p.127-62.
14. Suter PM, Fairley HB, Isemberg MD, et al. Optimum end-expiratory airway pressure in patients with acute pulmonary failure. N Engl J Med 1975;292:284-9.
15. Gattinoni L, Pelosi P, Vitale G, et al. Body position changes lung computed tomographic density in patients with acute respiratory failure. Anesthesiology 1991;74:15-23.
16. Broccard AF, Shapiro RS, Schmitz LL, et al. Influence
of prone position on the extent and distribution of lung injury in high volume
oleic acid model of acute respiratory distress syndrome. Crit Care Med
1997;25:16-22.